一种遗传性凝血功能异常的出血性疾病
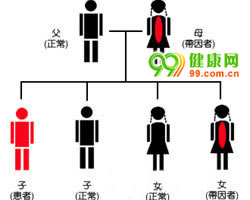
血友病属于一种X染色体连锁的隐性遗传性出血性疾病,是由于体内凝血因子Ⅷ(FⅧ)基因或凝血因子Ⅸ(FⅨ)基因缺陷,导致FⅧ或FⅨ缺乏,而使患者终身凝血功能异常,终身易于出血。
血友病是一种遗传性出血性疾病,自出生时即可发病,伴随终身。如患者得不到标准规范的治疗,有很高的致残及致死率。而接受标准规范治疗的患者,可以相对正常的生长发育,避免残疾。在发达国家,血友病患者的生活质量及预期寿命基本接近正常人群。
有关血友病的最初记载在公元二世纪的犹太法典中出现。19世纪,血友病的携带者通过王室间的通婚使血友病在多个欧洲王室中出现。现在我们已知,在欧洲王室中流行的是血友病B。
血友病绝大多数患者为男性,女性血友病患者罕见。
血友病的发病率没有地区和种族差异,在男性人群中,血友病A的发病率为1/5000,血友病B的发病率为1/25000。女性血友病患者罕见。
血友病的患病率,由于经济等各方面的原因,在不同国家甚至同一国家的不同时期都存在很大的差异。1986至1989年期间在全国24个省的37个地区进行的调查结果显示,我国血友病的患病率为 2.73/10万人口。按此患病率推算,目前我国血友病患者总数约为4万左右。
根据患者缺乏凝血因子的类型,可以分为:血友病A或血友病甲(FⅧ缺乏)及血友病B或血友病乙(FⅨ缺乏)。历史上曾经把FⅪ缺乏称为血友病C或血友病丙,但是由于FⅪ的遗传方式与血友病A或B截然不同,因此现在并不用此名称。
所有血友病患者中,血友病A占80%~85%,血友病B占15%~20%。女性血友病患者罕见。
根据患者体内残存的FⅧ活性(FⅧ:C)及FⅨ活性(FⅨ:C),血友病可以分为:重型(活性<1%)、中间型(1%~5%)及轻型(5%~40%),不同因子活性程度的患者具有不同的出血表现,也是决定患者需要采取何种治疗方案的基础。